Research Scientific achievements and research interests
International Mouse Phenotyping Consortium (IMPC)
We have been involved in the IMPC (Groza et al., 2023) since its inception.
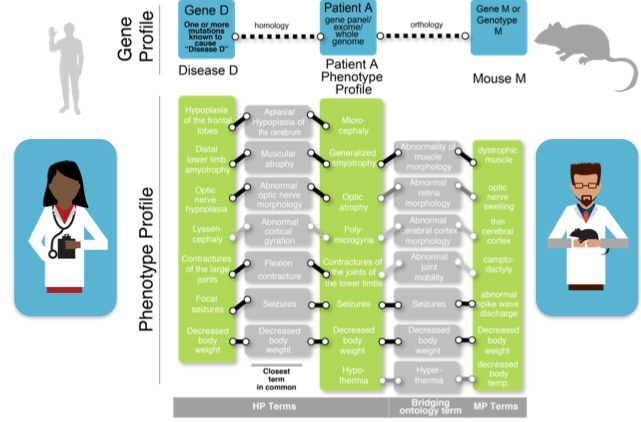
Our team analyses the genotype to phenotype associations emerging from this comprehensive effort to produce the first catalogue of mammalian gene function by knocking out and systematically phenotyping every protein-coding gene in the mouse. Utilising phenotype comparison methods developed with his co-PIs in the Monarch Initiative, his team is able to automatically identify new animal models of known disease genes as well as suggest new candidates for diseases where the causative variants have not yet been identified in human (Meehan et al., 2017). We also integrate IMPC viability data with other gene essentiality sources to predict new disease genes (Cacheiro et al., 2020).
The Monarch Initiative
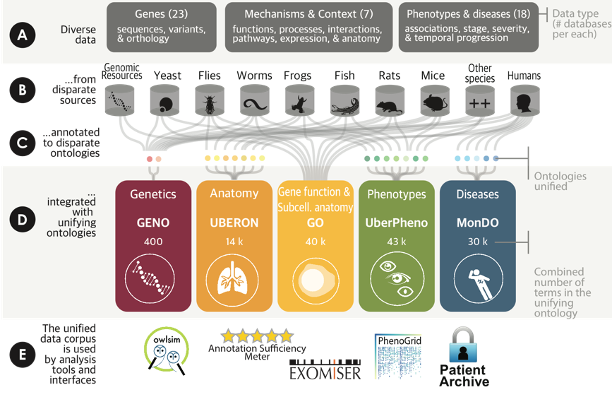
The Monarch Initiative is an integrative data and analytic platform connecting phenotypes to genotypes across species, bridging basic and applied research with semantics-based analysis. The correlation of phenotypic outcomes and disease with genetic variation and environmental factors is a core pursuit in biology and biomedicine. We have created or currently contribute to many essential bio-ontologies that together enable sophisticated and semantically integrated computational analysis across gene, genotype, variant, disease, and phenotype data. We have developed algorithms and tools that are in use by multiple communities for tasks including the identification of animal models of human disease through phenotypic similarity, phenotype-driven computational support for differential diagnostics, and translational research.
The Monarch Initiative is an extensive knowledge graph and ecosystem of tools made for the benefit of clinicians, researchers, and scientists. The knowledge graph consists of millions of entities – genes, diseases, phenotypes, and many more – imported from dozens of sources.
Exomiser
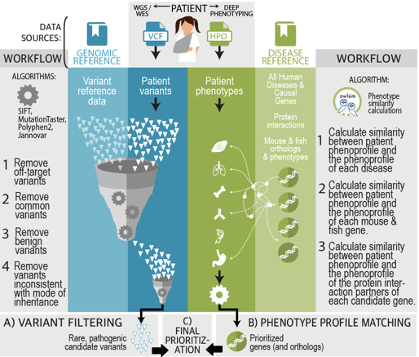
Exomiser automates the filtering and prioritisation of coding and non-coding variants called from whole exome or genome sequencing of rare disease families using novel methodologies to prioritise the genes based on the similarity of the patient’s phenotypes to reference knowledge of genotype to phenotype associations from human disease and animal models. This software is widely used by academic researchers, diagnostic laboratories, commercial offering and in large-scale disease sequencing projects such as the US Undiagnosed Disease Network, the UK’s 100,000 Genomes Project as well as being a key component of the ISO-accredited interpretation pipeline for the NHS Genomic Medicine Service.
The 100,000 Genomes Project

Prof. Smedley served as Director of Genomic Interpretation from 2016-2018 at Genomics England, helping to deliver the 100,000 Genomes Project. He was particularly involved in the rare disease component as described in Smedley et al., NEJM, 2021 and incorporating a phenotype-driven approach using Exomiser into this pipeline. We are now involved in various projects making use of the genomic and clinical data collected by the 100,000 Genomes Project to discover new disease genes and to find diagnoses in unsolved cases through novel strategies investigating reinterpretation of coding variants, characterisation of non-coding and structural variants, and use of other omic technologies.
Molecular Phenotypes of Null Alleles in Cells (MorPhiC)
The MorPhiC programme aims to develop a consistent catalog of molecular and cellular phenotypes for null alleles for every human gene by using in-vitro multicellular systems. The catalog will be made available for broad use by the biomedical community. MorPhiC has three components: the Data Production Research and Development Centers (DPCs), the Data Analysis and Validation Centers (DAVs) and the Data Resource and Administrative Coordinating Center (DRACC).We are part of the DRACC where amongst other activities we are building tools to help choose and characterise the genes that will be studied.